Run HSDFinder
Visualization
To comparative analyze the HSDs across different species, we developed an online heat map plotting option to visualize the HSDs results in different KEGG pathway category.
Create Heatmap [Download Hands-on protocol to create heatmap.pdf]
What's new
Aug. 5th, 2020: Updated to version 1.5.
The result of the predicted HSDs is displayed in a spreadsheet, which offers an alternative way to browse the result in graphical and tabular form.
The software presented here is the primary selection of HSDs, the manually curation should be done to filter the partial and pseudogenes.
Aug. 1st, 2020: Updated to version 1.0.
The web server is able to analyze the unannotated genome sequences by integrating the results from InterProScan (e.g., Pfam) and KEGG.
What's HSDatabase
HSDs has served as a critical database in the eukaryotic genomics, and the database has facilitated the studies associated with the highly similar duplcaites, such as gene duplicates detections, gene duplicates prediction. The predictions of HSDs stored in HSDatabse have been supported by a series of experiments, including those published in New Phytologist, iScience etc.. The current web is available at HSDatabase.
Tutorial
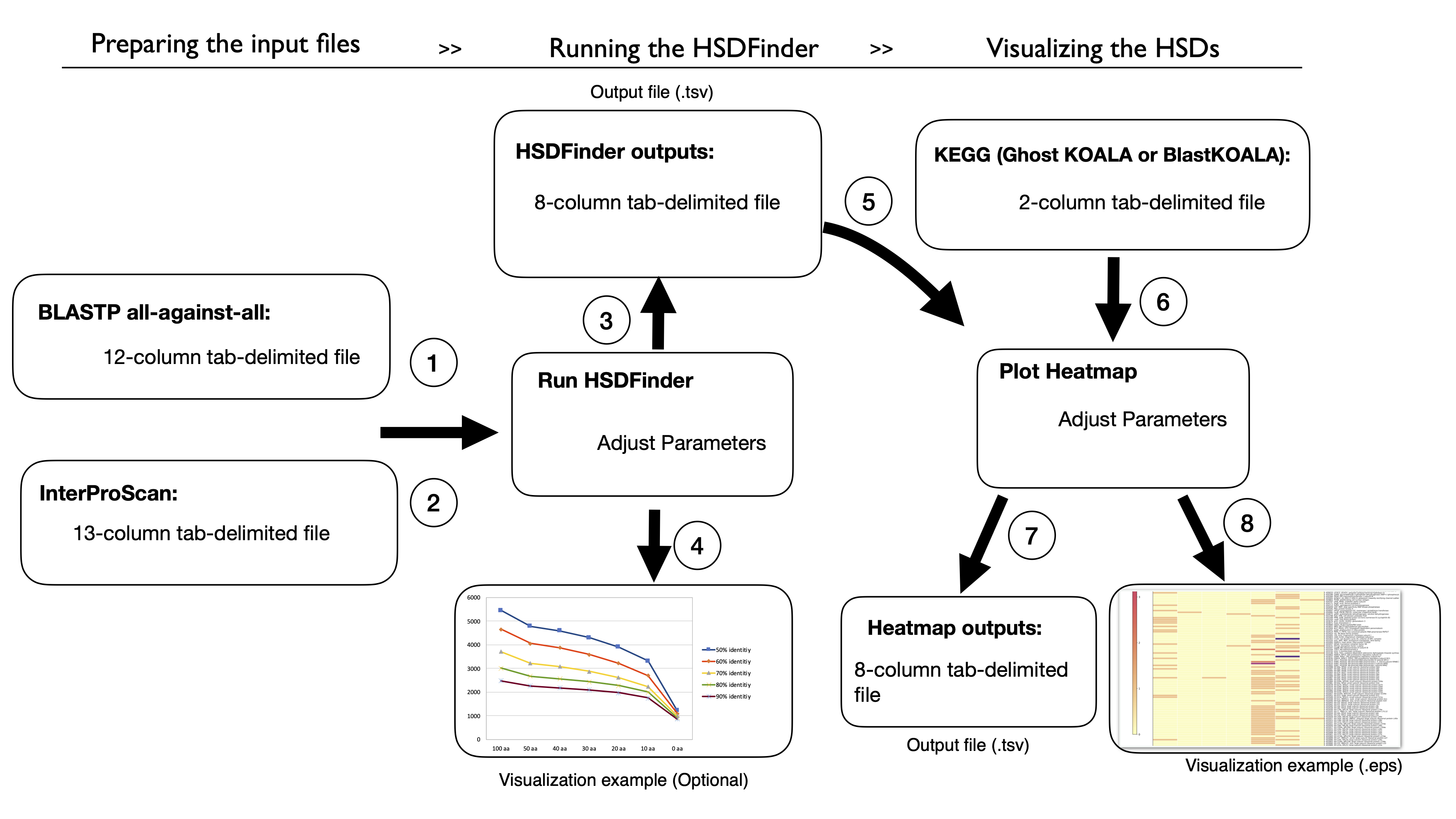
Workflow of HSDFinder (8 steps) [Download Tutorial for HSDFinder.pdf]
HSDFinder Online server Tutorial: Identification of highly similar duplicates in eukaryotic genomes.
1. Upload a protein BLAST search result file of your genome in tab-separated values (tsv) format as the first input file (File 1) of HSDFinder.
2. Upload a InterProScan search result file of your genome in tab-separated values (tsv) format as the second input file (File 2) of HSDFinder.
3. Yielding the output of HSDFinder with three personalized options.
4. Visualizing the HSDFinder outputs via the Excel tools (optional).
5. Upload the results of HSDFinder from your respective genomes.
6. Upload a gene list with KO annotation from KEGG database.
7. The output files of the online Heatmap Visualization tool.
8. The heatmap of HSDs levels across species.
Webinars: Using Bioinformatics Tools to Predict, Collect and Visualize the Highly Similar Duplicates in Eukaryotic Genomes
HSDFinder - an integrated tool to predict highly similar duplicates (HSDs) in eukaryotic genomes. (http://hsdfinder.com)
Series 1: How to run HSDFinder?
Series 2: How to visualize HSDs in HSDFinder?
Series 3: How to run HSDFinder locally by downloading from GitHub?
HSDatabase - a database of highly similar duplicate genes in eukaryotic genomes. (http://hsdfinder.com/database/)
Series 4: How to use the HSDatabase - browse and search?
Series 5: How to use the HSDatabase - BLAST and KEGG?
How to cite
Xi Zhang, Yining Hu, Zhengyu Cheng, John M. Archibald (2023). HSDecipher: A pipeline for comparative genomic analysis of highly similar duplicate genes in eukaryotic genomes. Star Protocols. doi: https://doi.org/10.1016/j.xpro.2022.102014
Xi Zhang, Yining Hu, David Roy Smith (2022). An overview of online resources for intra-species detection of gene duplications. Frontiers in Genetics. doi: http://doi.org/10.3389/fgene.2022.1012788.
Xi Zhang, Yining Hu, David Roy Smith (2022). HSDatabase - a database of highly similar duplicate genes from plants, animals, and algae. Database. doi: http://doi.org/10.1093/database/baac086.
Xi Zhang, Yining Hu, David Roy Smith (2021). HSDFinder: a BLAST-based strategy to search for highly similar duplicated genes in eukaryotic genomes. Frontiers in Bioinformatics. doi: http://doi.org/10.3389/fbinf.2021.803176
Xi Zhang, Yining Hu, David Roy Smith. (2021). Protocol for HSDFinder: Identifying, annotating, categorizing, and visualizing duplicated genes in eukaryotic genomes. STAR Protocols. DOI:https://doi.org/10.1016/j.xpro.2021.100619
Xi Zhang, Marina Cvetkovska, Rachael Morgan-Kiss, Norman P. A. Hüner, David Roy Smith, (2021). Draft genome sequence of the Antarctic green alga Chlamydomonas sp. UWO241. iScience. https://doi.org/10.1016/j.isci.2021.102084
Where to download
The distribution version of HSDFinder is also available.
Current version: v1 (5 August 2020) [download].
Links to the InterProScan and KEGG
InterProscan: https://github.com/ebi-pf-team/interproscan
KEGG : https://www.kegg.jp/kegg/
Frequently Asked Questions (FAQs)
awk '/^>/{if (l!="") print l; print; l=0; next}{l+=length($0)}END{print l}' '/.../.../protein.fa' |paste - - |sed 's/>//g'|awk -F'\t' '{print $1"\t"$1"\t"100"\t"$2}' >##.protein.length.aa This output file "##.protein.length.aa" can simply paste into the "##.BLAST.tabular" to run as the input file.